


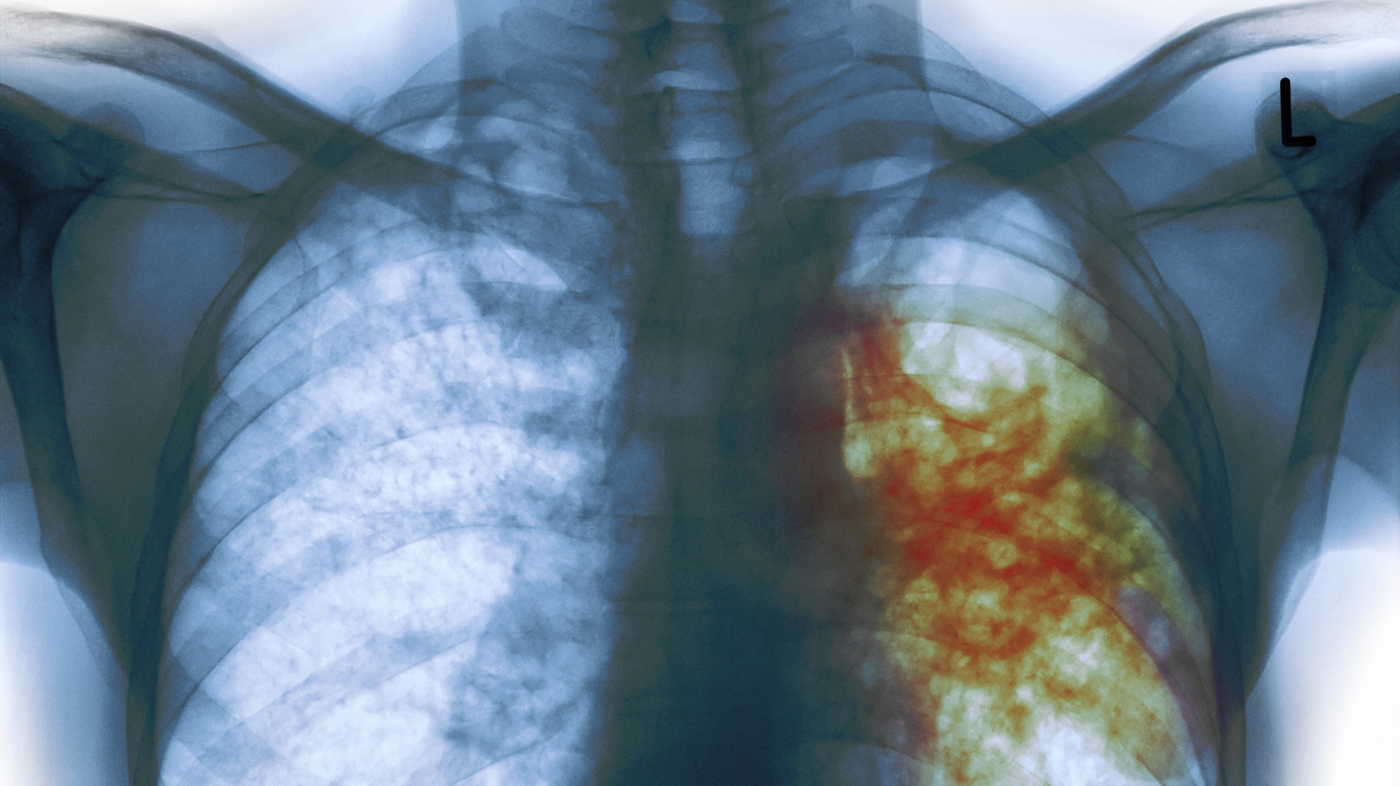
Tuberculosis (TB), caused by the pathogen Mycobacterium tuberculosis, is a serious global health problem accounting for 1.3 million worldwide deaths annually. There is no vaccine for TB, and treatment is a 6-9 month course of multiple antibiotics, which results in high rates of non-compliance and the emergence of drug-resistant strains. Scientists funded by the National Institute of Biomedical Imaging and Bioengineering used computers to model the formation of tuberculosis granulomas in the lung — the non-active (latent) form of infection found in 2 billion individuals worldwide (11 million in the U.S.) that can activate to become a life-threatening infection. Employing a computer model aims to speed analysis of TB’s complex life-cycle and to identify potential new antibiotics, antibiotic targets, and biomarkers that can predict transition to active infection.
TB granuloma form in the lungs when an individual inhales the bacterium. The granular structure develops as immune cells attack but are unable to destroy the bacterium. Instead, the bacteria enter and reside in the immune cells—specifically, macrophages–forming the center of the granuloma. A large number of uninfected macrophages are attracted to the site, surrounding the central infected cells. The 1-3 millimeter granuloma also contains fibrous and necrotic tissue as well as a number of other types of immune cells, making it an extremely complex structure.
Battling an infection harbored in one third of the world’s population
For Denise Kirschner, Ph.D., Co-Director of the Center for Systems Biology at the University of Michigan, and senior leader of the group, TB research centers on determining what makes a single granuloma one that will remain latent versus one that will become uncontrolled and spread numerous deadly bacteria.
“Discovering the factors involved in granuloma control vs those not able to control bacterial growth is a critical step in potentially developing treatments that could “freeze” granulomas into a protective state in the two billion latent carriers,” explains Grace Peng, Ph.D., Director of the NIBIB program for Mathematical Modeling, Simulation and Analysis. “Computer modeling, also known as in silico modeling, of such a complex disease allows us to work toward potential treatments much more rapidly.” The work is reported in two articles in the April issues of PLOS Computational Biology, and Infection and Immunity.
Kirschner and her colleagues have turned to developing and studying computer models because there are numerous interacting variables in the process of infection that include tissues, cell types (both lung and immune cells), growth factors, and molecules involved in immune cell signaling such as cytokines and interferons. The research approach is called multi-scale modeling or MSM, to reflect the ability to study disease processes at the molecular, cellular, and tissue levels as well as interactions occurring between the levels. Kirschner’s project is part of the Multiscale Modeling Consortium, coordinated by the Interagency Modeling and Analysis Group (IMAG).
Although there are animal models of TB infection, results can be slow to emerge and also can be costly. For example, it takes on the order of months to establish a TB infection in non-human primates (NHPs). However, animal results are a critical measure of the validity of the computer models being developed. All the characteristics and aspects of TB infection and granuloma formation, gained from years of research in animals, are the basis for constructing the computer model of TB infection.
Kirschner explains that they run computer simulations of infection and granuloma formation to see what happens when some factors are removed or a drug intervention is added to the model. “We then compare the results obtained from the computer model with results of our collaborators doing more laborious experiments in rabbits, mice, or NHPs. As a way to validate the models, if our results are similar to those observed in animals, we know the model is on the right track and is replicating what happens in the host. If not, we will adjust the inputs into the model in an effort to make it as similar as possible to what is observed in animals.” This is a highly iterative process, according to Kirschner.
The researchers make changes in the model such as computationally adding a potential drug or virtually knocking out the gene for a molecule produced by the immune system to search for changes that cause the granuloma to move toward a controlled state. The results could aid discovery of a drug that renders the TB granuloma better able to control bacterial growth.
Searching for biomarkers that predict granuloma behavior
In addition to looking for potential drugs and drug targets to control infection, the group is also addressing a major hurdle in controlling TB, which is the lack of accurate biomarkers that can predict granuloma activation. This is extremely important given the enormous latent human reservoir for potential disease transmission. As Kirschner puts it, “An individual may have hundreds of latent granulomas, but if just one becomes activated it could spread in the lungs and without treatment lead to death or transmission of infection to others.”
Therefore, a crucial endeavor of the group is to identify biomarkers –molecular or cellular changes that can be measured fairly easily from blood– that would provide accurate information to predict whether granulomas are likely to remain latent or transition to active infection.
The community of TB researchers agrees that such biomarkers are one of the greatest tools in disease diagnosis and treatment, but there is debate about whether such markers can be found in the context of the complex cellular and molecular environment created by a TB infection.
Employing large in silico data sets to identify useful biomarkers
To tackle the challenge of identifying valid biomarkers the group created a multi-organ, computational model that tracks infection dynamics in lungs, blood, and lymph nodes using the extensive non-human primate datasets that were available. This approach allowed for the creation of thousands of in silico datasets to mine for potential biomarkers.
Their preliminary results identified two types of immune cells, CD4+ and CD8+ T cells, which become mobilized to attack various surface molecules on the TB bacterium. Coupled with the fact that replicating and non-replicating bacteria have different cell surface proteins, the initial experiments suggest that a biomarker, easily assayed in blood samples, could be developed based on the appearance of CD4+ and CD8+ T cells specific for TB bacteria.
The researchers are optimistic that they have identified a very promising approach for establishing reliable biomarkers. Current efforts are focused on identifying TB-specific surface proteins that are targeted by CD4+ and CD8+ T cells and testing them in their multi-organ computational model. The hope is that this iterative process will eventually winnow down those specific immune cells that are present in the blood of hosts during either latent or active infections. Such a blood test would be easy to use in all settings including rural, low resource regions.
Concludes Peng, “given the ubiquitous and complex nature of tuberculosis, it is fortunate that the Kirschner group’s work is rapidly advancing our understanding of the mycobacterium’s interaction with the human host. This appears to be a case where solving the complexity of the organism requires a partnership between clinical observations, animal studies, and the elegant computer models that the group continues to refine.”
Source: National Institute of Biomedical Imaging and Bioengineering









